Help
- About the server
- Navigation bar
- Input of Fast Search of Structure Database
- Input of Multiple Protein Structure Alignment
- Output of Fast Search of Structure Database
- Output of Multiple Protein Structure Alignment
- Output of Pairwise Protein Structure Alignment
- Reference
About the server
The server consists of two related modules: fast search of structure database and multiple protein structure alignment. The structure search could be performed on different databases include PDB, SCOPe, CATH, BFVD, and AlphaFoldDB. The multiple protein structure alignment is performed using the fast and accurate algorithm mTM-align.
The database search consists of two steps:
The multiple structure alignment is built with three steps:
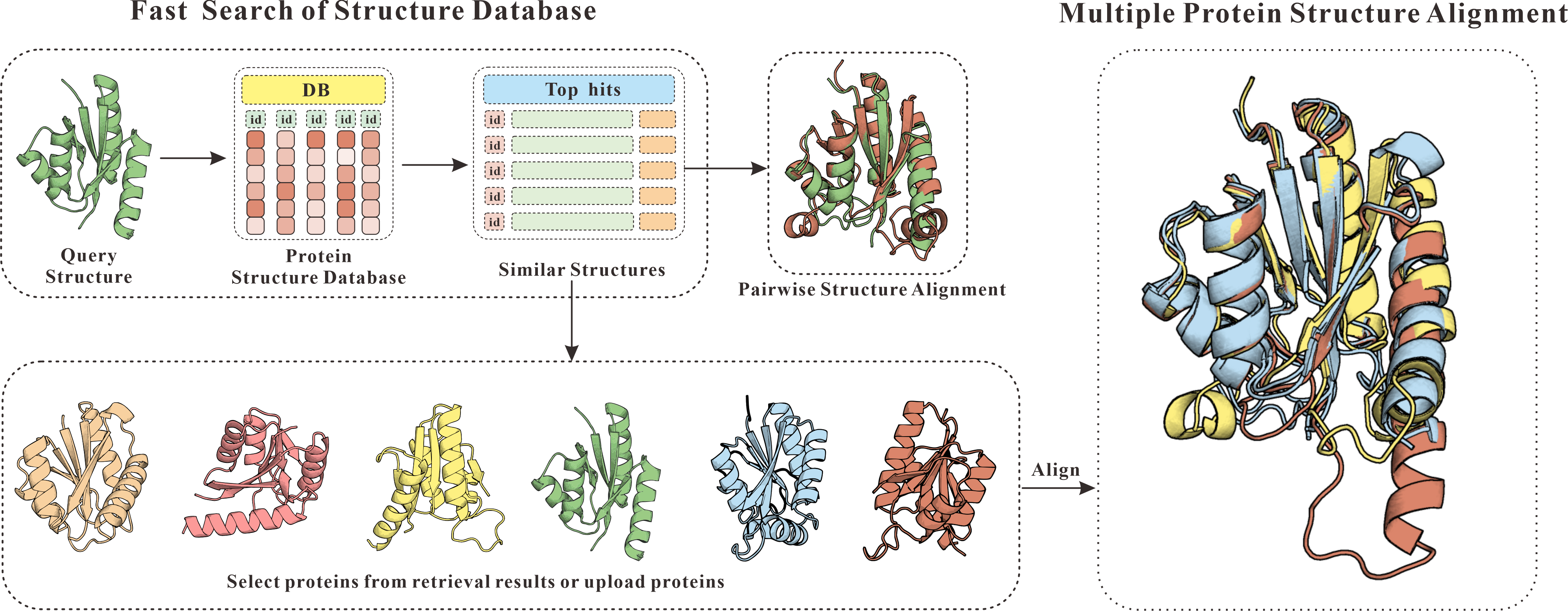
Figure 1. The flowchart of mTM-align server.
The navigation bar includes links to the main page of Yang Lab (①) and the mTM-align server (②). Additionally, it offers examples (③④) demonstrating how our server operates, along with hits from various benchmark (⑤) tests. News (⑥) for mTM-algin are also provided.
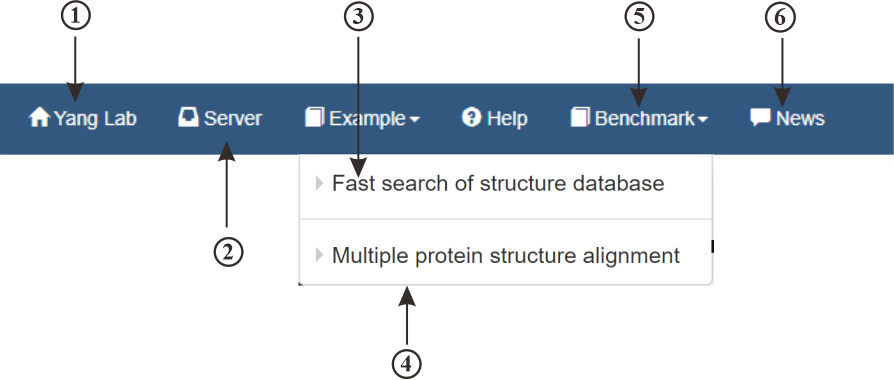
Figure 2. The navigation bar of mTM-align server.
Input of Fast Search of Structure Database
This (①) is the input page for retrieving protein structure data. You can test our service using the provided example (②③). You have the option to input your protein sequence directly into the text box (④), upload a protein file (⑤), or load a protein (⑥) from the Protein Data Bank (PDB) or AlphaFold Database (AFDB). You can select the specific database (⑦) you wish to query and choose the retrieval mode (⑧) that best suits your needs. Additionally, you can provide your email address (⑨) to receive the retrieval results directly.
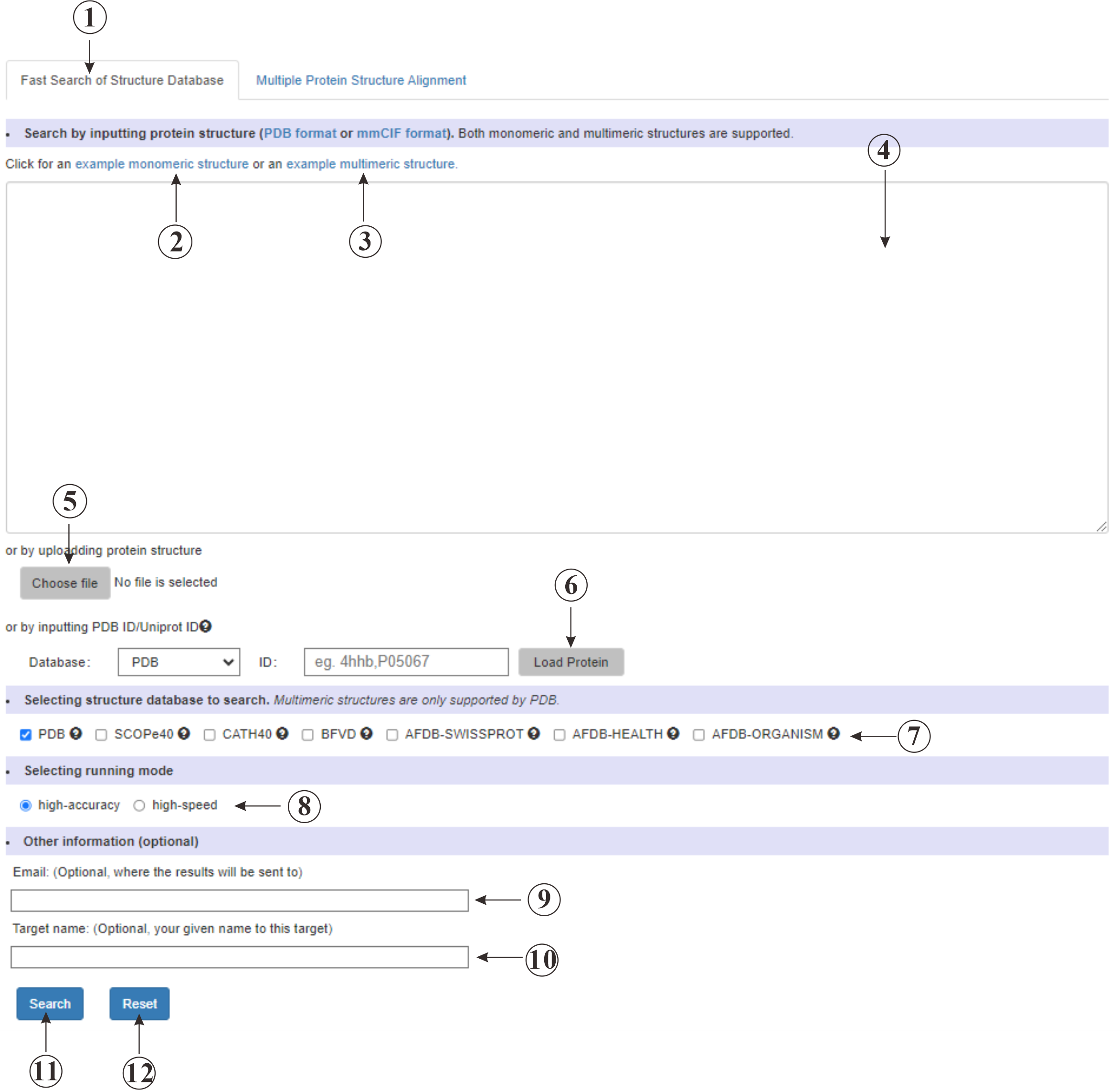
Figure 3. The submssion page of Fast Search of Structure Database.
Input of Multiple Protein Structure Alignment
This (①) is the input page for performing multiple protein structure alignment. You can download an example zip file(②) to validate our service. You can also upload (③) a compressed file containing multiple protein structures. Up to 20 structures are accepted.
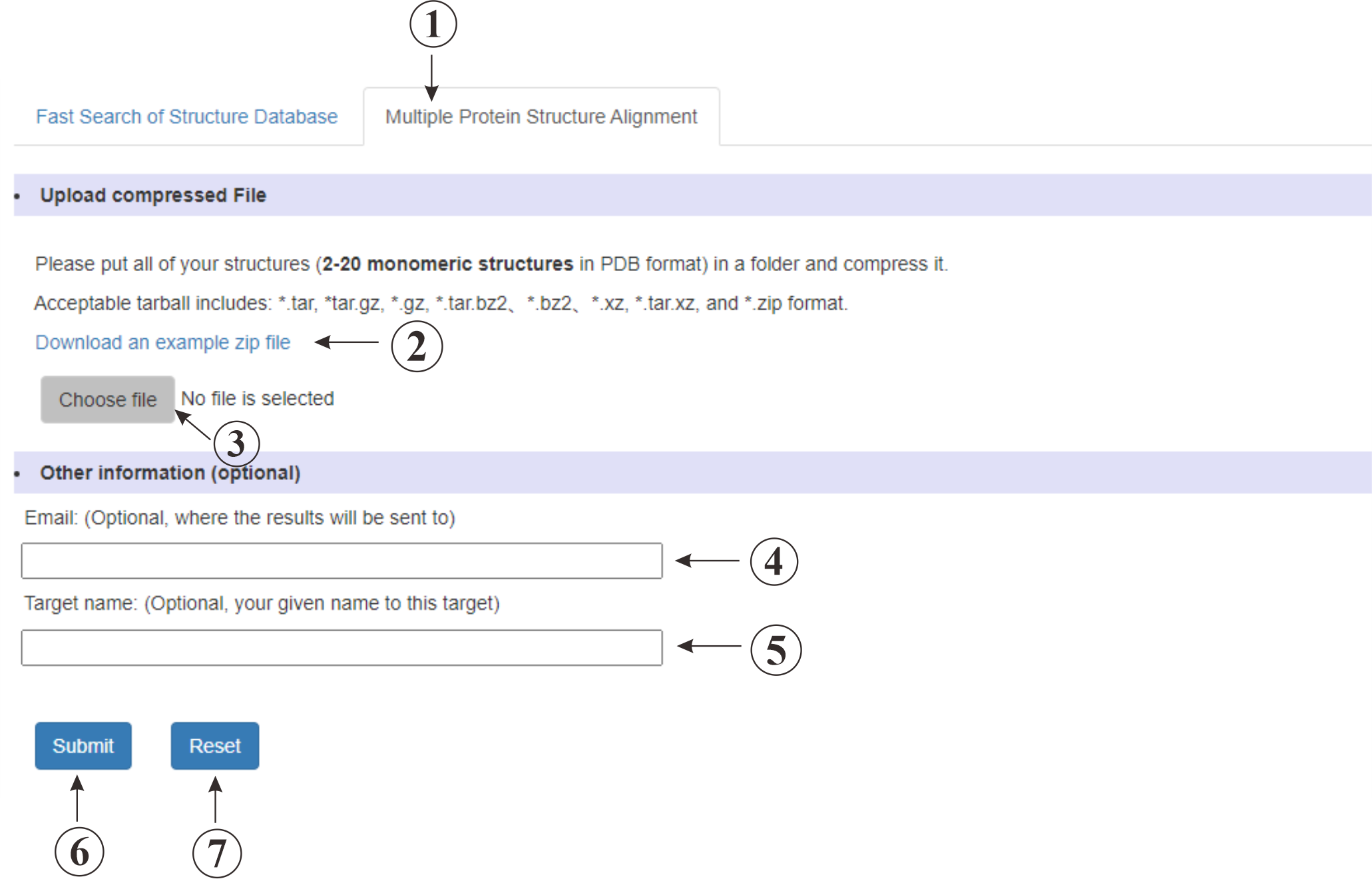
Figure 4. The submssion page of Multiple Protein Structure Alignment.
Output of Fast Search of Structure Database
This is the result page for retrieving protein structure data. The page displays proteins that share similar structures with your query. The organism (⑪) and molecule (⑩) information are also provided. You have the option to download (⑥) these proteins or align them with your query protein. Both pairwise (⑦) and multiple (④) structure alignments are supported. For multiple structure alignment, you should click on the checkbox (⑨) to choose the proteins that you want to align. Maybe you could click on ① to acquire the alignment for the top 10 result in this DB. Proteins from different DBs (③) are also accepted. Additionally, we offer prediction of the query's binding sites with the result structures. Please click on align (⑦) button to view the prediction.
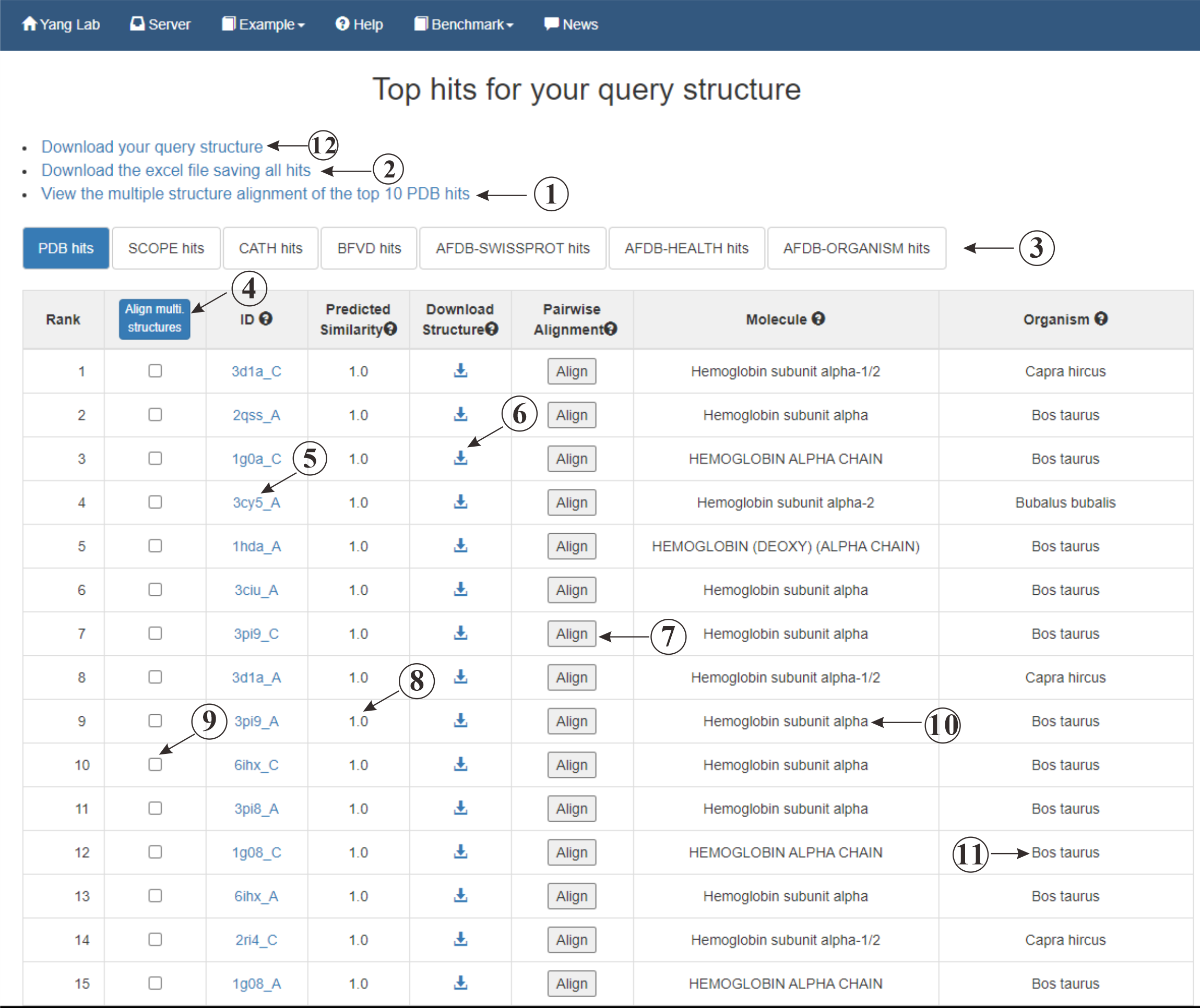
Figure 5. The output of Fast Search of Structure Database.
Output of Multiple Protein Structure Alignment
This is the result page for multiple protein structure alignment. We offer a visualization (①) for the aligned proteins, various viewing options (⑩⑪⑫) are provided. The alignment is also displayed at the residue level (②), and we provide a structure-based phylogenetic tree (③) for further analysis. Several indicators (④⑤⑥⑦⑧⑨) are shown to help you better understand the similarities between the proteins. Additionally, you can download (⑬) the files for the common core regions and the whole proteins.
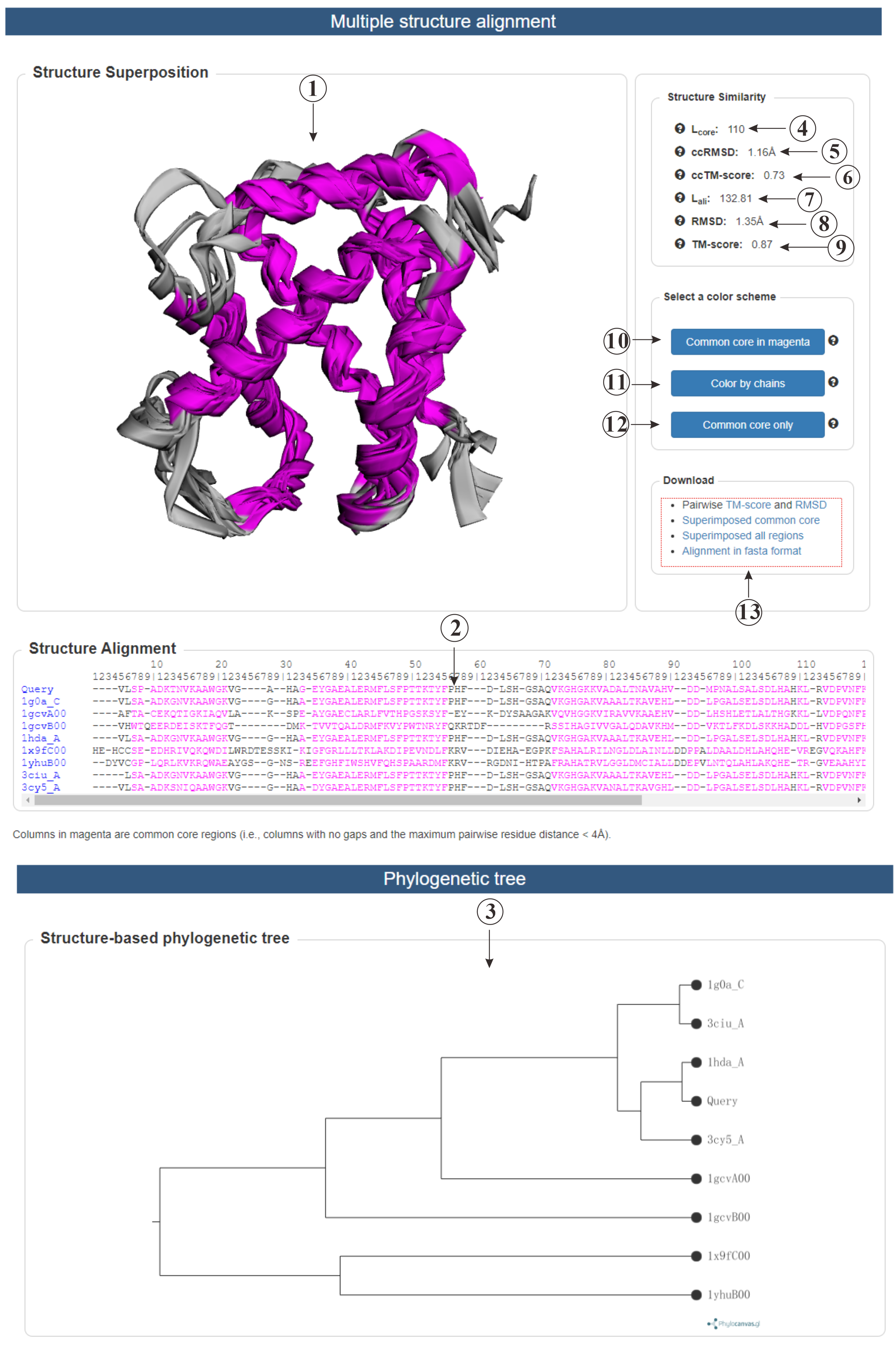
Figure 6. The output of Multiple Protein Structure Alignment.
Output of Pairwise Protein Structure Alignment
This is the result page for pairwise protein structure alignment. Firstly, we provide a visualization (①) of the aligned proteins, along with a residue-level structure alignment (⑫). Secondly, we predict the ligand (⑩) of the query protein based on the template. Clicking on the corresponding button to show the ligand along with the predicted binding residues (⑪). Additionally, various indicators (②③④⑤⑥⑦⑧⑨) are included to help understanding the similarity of the two proteins.
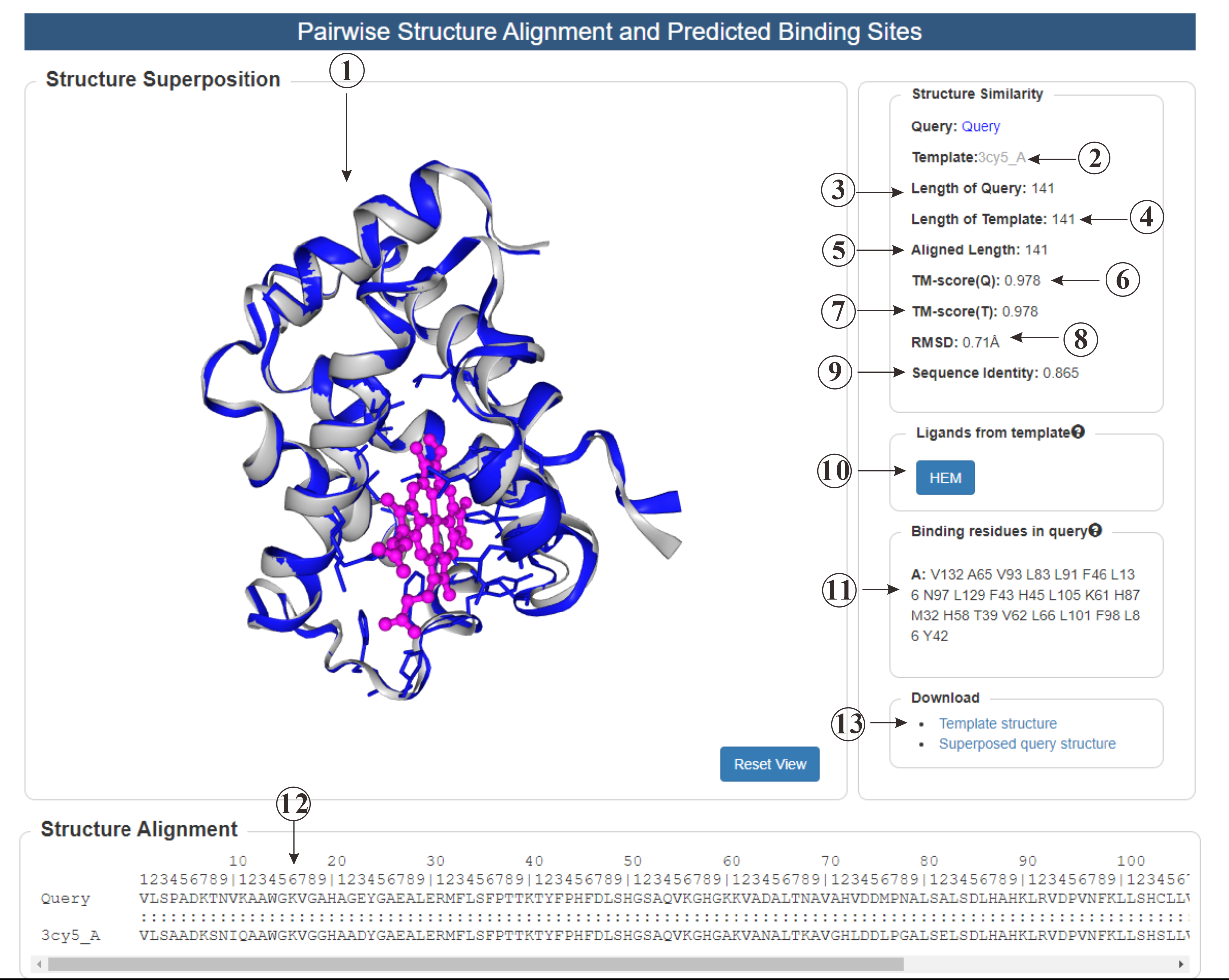
Figure 7. The output of Pairwise Protein Structure Alignment.
References