![]()
Help
- About the server
- Navigation bar
- Input of Fast Search of Structure Database
- Domain split
- Input of Multiple Protein Structure Alignment
- Output of Fast Search of Structure Database
- Output of Multiple Protein Structure Alignment
- The difference in the performance of search with the chain and the domain databases
- Reference
About the server
The server consists of two related modules: fast search of structure database and multiple protein structure alignment. The database search is speeded up based on a heuristic algorithm and a hierarchical organization the structures in the database. The multiple protein structure alignment is performed using the fast and accurate algorithm mTM-align.
The database search consists of three steps:
The multiple structure alignment is built with three steps:
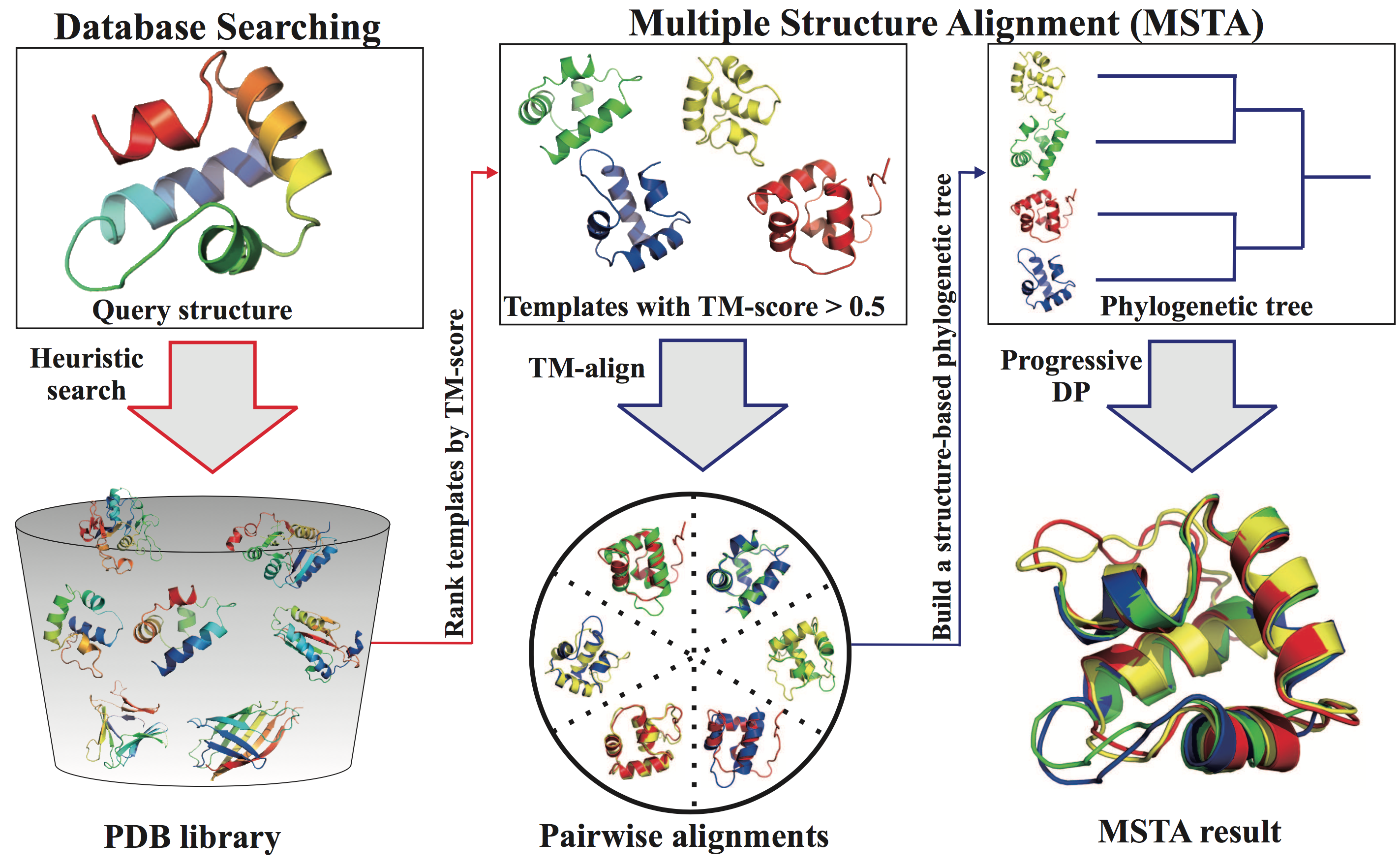
Figure 1. The flowchart of mTM-align server.
Navigation bar

Figure 2. The Navigation bar.
Input of Fast Search of Structure Database
For fast database search, you are allowed to either paste your structure into text box, or upload your structure as a PDB file (Figure 3). If you choose "Advanced Option" and select 'Yes' for the option "Split or not", your structure will be cut into domains by PDP (protein domain parser).

Figure 3. The submssion page of Fast Search of Structure Database.
Domain Split
You can choose one of them to start search (Figure 4).

Figure 4. The domain selection page of Fast Search of Structure Database.
Input of Multiple Protein Structure Alignment
For Multiple Protein Structure Alignment, you should upload a tarball with at least two structures in it (Figure 5).

Figure 5. The submssion page of Multiple Protein Structure Alignment.
Output of Fast Search of Structure Database
The output of Fast Search of Structure Database is a list of templates (Figure 6). You can click on "View the automated MSTA with the top 10 templates" to view the alignment built with the top 10 templates. You can click on the template name to download the PDB file of each template or click the "link" to the link in the RCSB PDB website. In addition, you are also able to select the templates that you are interested in to perform multiple structure alignment.

Figure 6. The search result page of Fast Search of Structure Database.
Output of Multiple Protein Structure Alignment
The output of Multiple Protein Structure Alignment is shown in Figure 7. You can view the alignment or download it in fasta format. For visualization of the alignment, you can choose to view all structures or only the common core region.

Figure 7. The result page of Multiple Protein Structure Alignment.
The Difference in the Performance of Search with the Chain and the Domain Databases
The difference in performance of search with the chain and the domain databases is shown in Figure 8, assessed on a dataset of 500 structures from the SCOPe database. A structure in the top n of the result list is defined as a true positive (TP) if its fold definition SCOPe is the same as the query. The mean precision (p(n)) and recall (r(n)) are used to measure the difference in performance of search with the chain and the domain databases.

Figure 8. The difference in the performance of the search against the chain (PDBC) and the domain (DOM) databases.
Reference
Need more help?
If you have more questions or comments about the server, please email yangjy![]() nankai.edu.cn.
nankai.edu.cn.